Tratamiento actual de la hemofilia

Se están desarrollando tratamientos para la hemofilia2,3
Las opciones de tratamiento para la hemofilia se han ampliado en los últimos años1 y siguen avanzando.2,3
Muchas personas con hemofilia necesitan perfusiones periódicas de factores de sustitución.1 Esto se debe a que las personas con hemofilia moderada o grave presentan niveles de factor de entre el 1% y el 5%, a diferencia de los niveles de entre el 40% y el 150% que presentan las personas sin hemofilia.4-7 Para mantener unos niveles de factores casi normales, las personas con hemofilia necesitan perfusiones con factores de sustitución. Dependiendo de los niveles de factores, algunos necesitan perfusiones intravenosas con una frecuencia de cada dos días.1
Los tratamientos para la hemofilia han ido mejorando, por lo que ahora son más eficaces para controlar los síntomas y reducir la frecuencia del tratamiento. Por ejemplo, se han introducido productos de semivida prolongada (SVP) que requieren menos perfusiones.1
Opciones de tratamiento disponibles actualmente





How Haemophilia Treatments Work
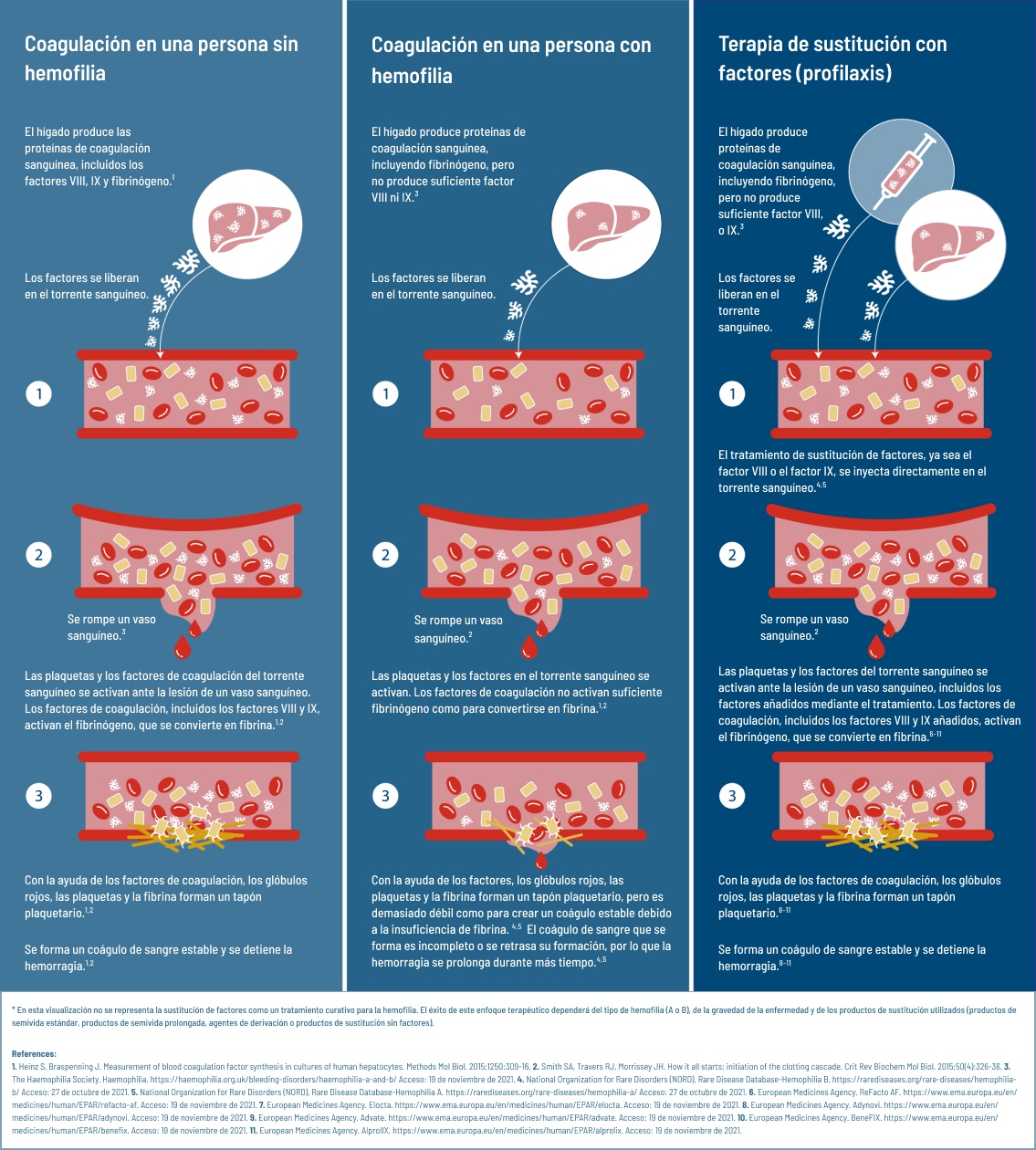
Factores sustitutivos: ¿Cómo funcionan?
Las personas sin hemofilia presentan niveles de factores de coagulación del 40% al 150%. Este porcentaje es muy superior al de las personas con hemofilia moderada (1-5%) o grave (menos del 1%).4-7 Para asegurar que las personas con hemofilia cuentan con suficientes factores de coagulación para evitar las hemorragias, las personas con hemofilia A reciben perfusiones periódicas de factor de coagulación VIII6 funcional y, en el caso de la hemofilia B, de factor IX.5
Las células hepáticas son las encargadas de producir los factores de coagulación.15 Cuando las personas con hemofilia reciben un tratamiento de sustitución de factores, se añade directamente al torrente sanguíneo. Esto significa que tienen la capacidad de formar coágulos de sangre,5,6 pero sin la ayuda del hígado. La única forma de mantener la capacidad de coagulación es añadiendo continuamente más factor de coagulación al torrente sanguíneo. Por desgracia, los factores de coagulación no duran mucho tiempo. Estas proteínas se agotan cada 8-12 horas (factor VIII)16 o 18-24 horas (factor XI).1 Por eso las células hepáticas producen continuamente más factores de coagulación y las personas con hemofilia necesitan recibir factores de sustitución con frecuencia. La pauta de tratamiento para recibir factores de sustitución depende tanto del tipo de hemofilia (A o B) como de la gravedad de la misma (niveles de factor naturales).5,6
Desarrollo del tratamiento
Hoy en día se puede acceder a una gran variedad de tratamientos y se están desarrollando más2,3
Puede que algunos recuerden las limitadas opciones que tenían las personas con hemofilia hace tan solo unas décadas, o son conscientes de ello. Hasta mediados de los años 60, cuando la Dra. Judith Pool descubrió cómo combinar los factores de coagulación de varias donaciones de sangre, la esperanza de vida media de las personas hemofílicas era de tan solo 20 años.12
Desde entonces, el tratamiento se ha revolucionado.1 Hoy en día, las personas con hemofilia no dependen de las donaciones de sangre5,12 y los productos de semivida prolongada permiten una mayor libertad y flexibilidad en las pautas de tratamiento.16,17 Aunque el tratamiento ha avanzado mucho, hoy en día se siguen desarrollando nuevas estrategias de tratamiento2,3 con el objetivo de mejorar aún más la calidad de vida de los pacientes.18
Tratamiento a lo largo del tiempo
Antes de la era moderna de la hemofilia, no existían tratamientos fiables. Entonces las personas con hemofilia recibían perfusiones de sangre completa que no contaban con suficiente sustitución de factores durante los episodios hemorrágicos. Por suerte, los tratamientos han ido avanzado, poco a poco, hasta el punto de que la mayoría de las personas con hemofilia pueden llevar un estilo de vida más libre y activo.5
1964
Se descubrió un método para aislar factores de coagulación
La Dra. Judith Pool descubrió que el factor VIII, dañado en la hemofilia A, es el último componente del plasma en descongelarse tras su congelación. Este descubrimiento permitió aislar los factores de coagulación de varios donantes y combinarlos para transfundir concentraciones más altas de factores de coagulación.19 Por consiguiente, la esperanza de vida aumentó hasta los 24 años.12
1976
Se desarrollan factores de coagulación estables y concentrados
La Dra. Nilsson publicó, por primera vez, un método para producir factores de coagulación concentrados en forma de polvo. A partir de entonces, las personas con hemofilia podían conservar la medicación y administrarse los factores de coagulación en casa, reduciendo así el número de desplazamientos semanales al hospital.5,9
Años70-80s
Un período oscuro para la hemofilia: el suministro de sangre está contaminado
A principios de los 70, muchas personas quedaron expuestas a la hepatitis tras recibir transfusiones de concentrado de factores.5,20 A principios de los 80, se detectó el VIH en el suministro de sangre y,5,13 a la larga, el 40% de los estadounidenses con hemofilia fallecieron por complicaciones relacionadas con el síndrome de inmunodeficiencia adquirida (SIDA).21
1992 - 1999
La EMA y la FDA aprueban el primer producto de factor recombinante modificado genéticamente
La Agencia Europea de Medicamentos (EMA) y la Administración de Alimentos y Medicamentos (FDA) de Estados Unidos aprobaron el primer tratamiento de sustitución del factor IX modificado genéticamente (1997; 1992) y posteriormente del factor VIII (1999; 1997), respectivamente.5,10,11
2014 - 2016
2011 - 2017
Comienzan los ensayos de los primeros productos de terapia génica
En 2011 se publicaron los resultados del primer ensayo clínico de terapia génica con el factor IX y en 2017 los resultados de terapia génica con el factor VIII.3,14

Anterior
Vivir con hemofilia hoy en día
Los recientes avances científicos han revolucionado los tratamientos disponibles, aunque todavía hay margen de mejora para garantizar la máxima calidad de vida de los pacientes.
Siguiente
Terapia génica
Descubra los avances que los científicos han realizado en los últimos 50 años y que han permitido a muchos pacientes con enfermedades genéticas y trastornos crónicos acceder a la terapia génica.
References
- Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020;26 Suppl 6:1-158.
- Nathwani AC, Reiss UM, Tuddenham EG, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371(21):1994-2004.
- Rangarajan S, Walsh L, Lester W, et al. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N Engl J Med. 2017;377(26):2519-2530.
- Berntorp E, Fischer K, Hart DP, et al. Haemophilia. Nature Reviews Disease Primers. 2021;7(1):45.
- National Organization for Rare Disorders (NORD). Rare Disease Database – Hemophilia B. Acceso: 27 de octubre 2021. Disponible en: https://rarediseases.org/rare-diseases/hemophilia-b/.
- National Organization for Rare Disorders (NORD). Rare Disease Database – Hemophilia A. Acceso: 24 de noviembre 2021. Disponible en: https://rarediseases.org/rare-diseases/hemophilia-a/.
- Center UoRM. Factor VIII (Antihemophilia Factor A). Acceso: 30 de noviembre de 2021.
- Mancuso ME, Santagostino E. Outcome of Clinical Trials with New Extended Half-Life FVIII/IX Concentrates. Journal of Clinical Medicine. 2017;6(4).
- Morfini M. The History of Clotting Factor Concentrates Pharmacokinetics. Journal of Clinical Medicine. 2017;6(3).
- European Medicines Agency (EMA). BeneFIX. Acceso: 30 de noviembre de 2021 Disponible en: https://www.ema.europa.eu/en/medicines/human/EPAR/benefix.
- European Medicines Agency (EMA). ReFacto AF. Acceso: 30 de noviembre de 2021 Disponible en: https://www.ema.europa.eu/en/medicines/human/EPAR/refacto-af.
- Hemophilia Federation of America (HFA). Bleeding disorders historial timeline. Acceso: 10 de noviembre de 2021 Disponible en: https://www.hemophiliafed.org/updated-historical-timeline/.
- The National Academies of Sciences E, and Medicine. New measures needed to protect U.S. blood supply from future threats posed by infectious diseases. Washington DC, USA 1995. Acceso: 9 de noviembre de 2021. Disponible en: https://www8.nationalacademies.org/onpinews/newsitem.aspx?RecordID=4989.
- Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. New England Journal of Medicine. 2011;365(25):2357-2365.
- Heinz S, Braspenning J. Measurement of Blood Coagulation Factor Synthesis in Cultures of Human Hepatocytes. Methods in Molecular Biology. 2015;1250:309-316.
- Ay C, Feistritzer C, Rettl J, et al. Bleeding outcomes and factor utilization after switching to an extended half-life product for prophylaxis in haemophilia A in Austria. Scientific Reports. 2021;11(1):12967.
- Chhabra A, Spurden D, Fogarty PF, et al. Real-world outcomes associated with standard half-life and extended half-life factor replacement products for treatment of haemophilia A and B. Blood Coagul Fibrinolysis. 2020;31(3):186-192.
- Perrin GQ, Herzog RW, Markusic DM. Update on clinical gene therapy for hemophilia. Blood. 2019;133(5):407-414.
- Pool JG, Hershgold EJ, Pappenhagen AR. High-potency Antihaemophilic Factor Concentrate Prepared from Cryoglobulin Precipitate. Nature. 1964;203:312.
- Kasper CK, Kindgon HS, Hellerstein IJ. Hepatitis and Clotting-Factor Concentrates. JAMA. 1971;221(5).
- White GC. Hemophilia: an amazing 35-year journey from the depths of HIV to the threshold of cure. Trans Am Clin Climatol Assoc. 2010;121:61-75.
- European Medicines Agency (EMA). Elocta. Acceso: 30 de octubre de 2021. Disponible en: https://www.ema.europa.eu/en/medicines/human/EPAR/elocta.
- European Medicines Agency (EMA). Alprolix. Acceso: 5 de noviembre de 2021. Disponible en: https://www.ema.europa.eu/en/medicines/human/EPAR/alprolix.
¿Tiene una pregunta? Póngase en contacto con Información Médica
Gracias por su consulta.
En breve nos pondremos en contacto con usted.
